A leggere il titolo di questo articolo vi starete chiedendo di cosa voglio parlare. Ebbene, di certo non voglio discutere di fantasy, né di una città esotica e nemmeno di un nuovo pianeta di un sistema solare lontano anni luce dal nostro.
Niente di tutto questo.
Mi è venuta voglia di descrivere la chimica di una molecola il cui funzionamento mi ha divertito ed appassionato moltissimo quando studiavo la biochimica nell’ormai lontano 1989. Poi vi dirò perché mi è venuta questa voglia.
La nomenclatura
La ninidrina ha nome IUPAC 2,2-diidrossi-1,3-diossoidrindene che, per i non chimici, non risolve alcun mistero. Anzi, volendo, lo complica ancora di più perché introduce una complessità maggiore nel dare un nome a qualcosa usando delle regole molto lontane dalla vita quotidiana. Potremmo pure chiamarlo tirannosauro cityplex, ma cambiare etichetta non permette di comprendere nulla in merito all’oggetto di cui voglio discutere.
Allora andiamo con ordine.
Partiamo dal termine indene.
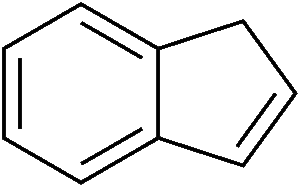
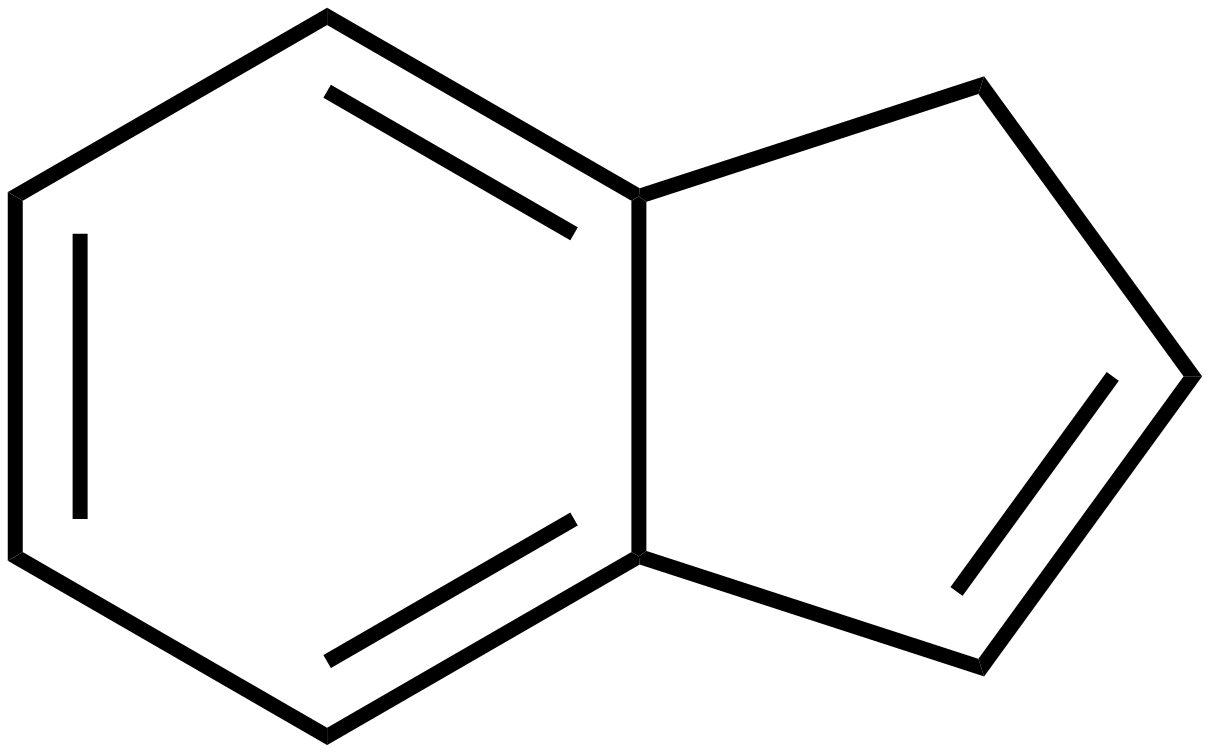
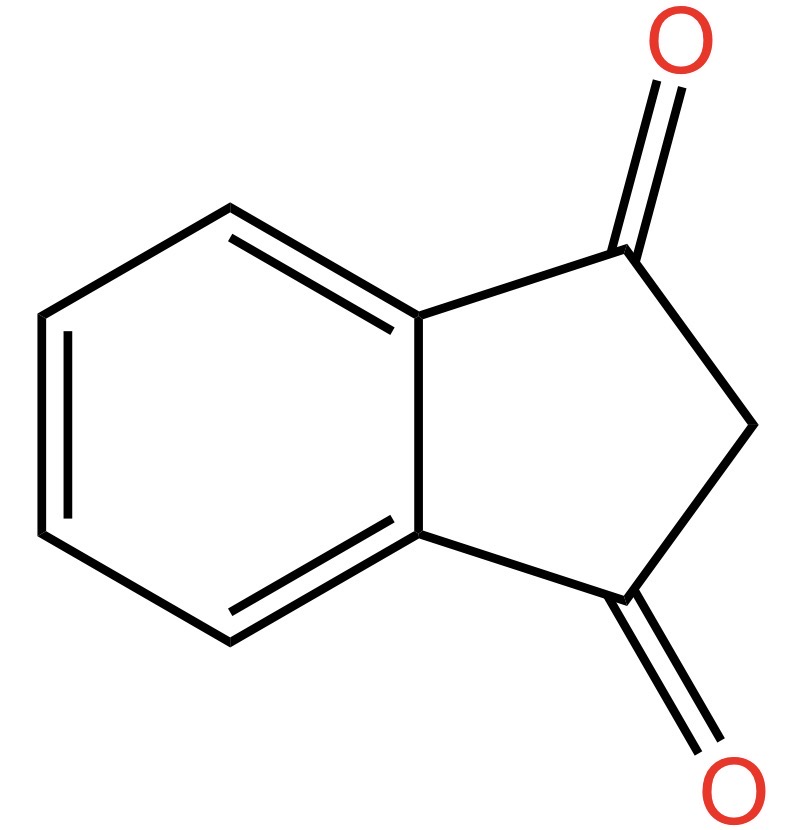
L’indene è la molecola descritta qui sotto. Come si vede, si tratta di un composto formato da un anello benzenico (quello a sinistra, a 6 atomi di carbonio) fuso con un anello ciclopentadienico (quello a destra, a 5 atomi di carbonio). L’anello a 5 termini contiene un doppio legame: quello che vedete in basso a destra. In realtà, i doppi legami nel ciclo a 5 atomi di carbonio sono due, ma non voglio complicarvi ulteriormente la vita con formule ed ibridi di risonanza, per cui decido scientemente di commettere un errore.
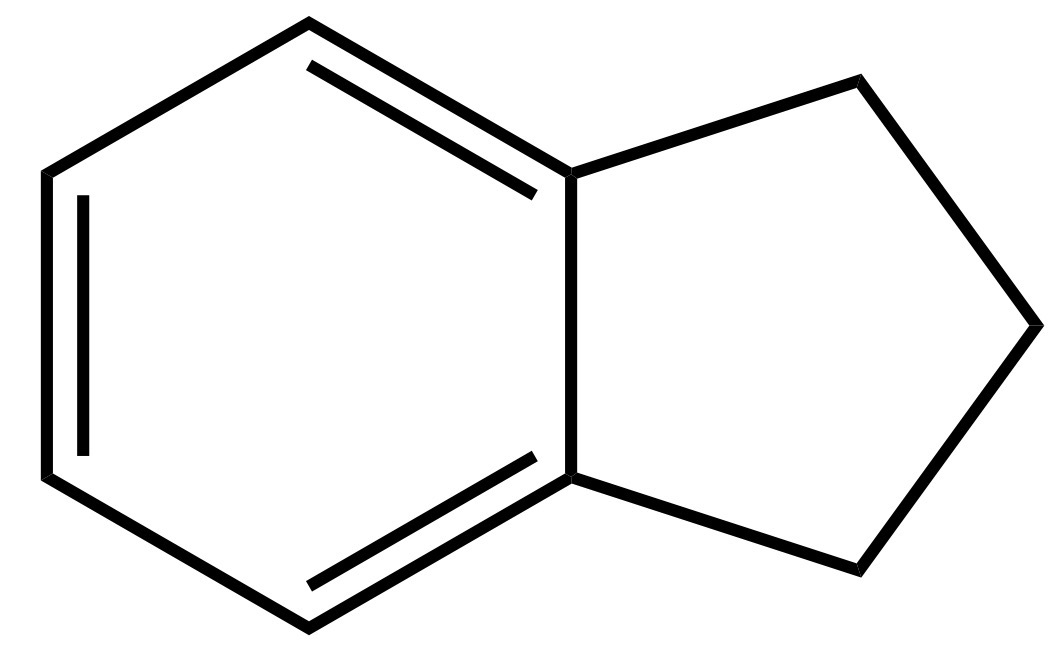
Se dall’anello a cinque atomi di carbonio togliamo quel doppio legame in basso a destra, otteniamo una molecola che si chiama idrindene la cui struttura è quindi:
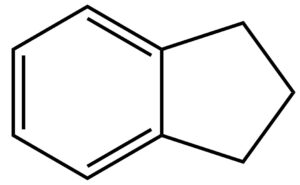
Il doppio legame lo potete immaginare come un tavolo in cui ci sono due posti vuoti. Ci siete voi che aspettate due vostri amici: i fratelli gemelli idrogeno1 e idrogeno2. Quando arrivano, si siedono e la tavola è completa. Da un punto di vista chimico, togliere un doppio legame significa inserire due atomi di idrogeno, uno su ogni atomo di carbonio coinvolto nel doppio legame, mediante una reazione che si chiama idrogenazione. Il suffisso “idr” in idrindene, quindi, si riferisce proprio all’indene idrogenato.
A questo punto mettiamo due atomi di ossigeno legati ciascuno con doppio legame agli atomi di carbonio 1 e 3 come indicato nella figura qui sotto.
Ricordate una cosa importante: in chimica organica vengono attribuiti i numeri civici agli atomi di carbonio presenti in una molecola. Questi numeri civici, che vengono indicati come posizione degli atomi di carbonio all’interno della molecola, servono ai chimici per comprendere di quale atomo nello specifico si stia parlando. A questo punto, scegliete voi quale dei due atomi di carbonio sia 1 e quale 3. Però tranquilli perché non c’è nessuna risposta giusta o sbagliata: la molecola è simmetrica; quindi, potete ribaltarla come in uno specchio e tutto funziona allo stesso modo.
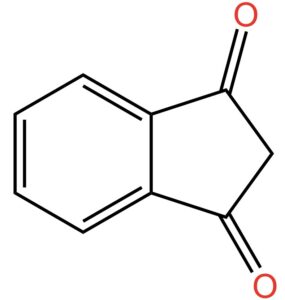
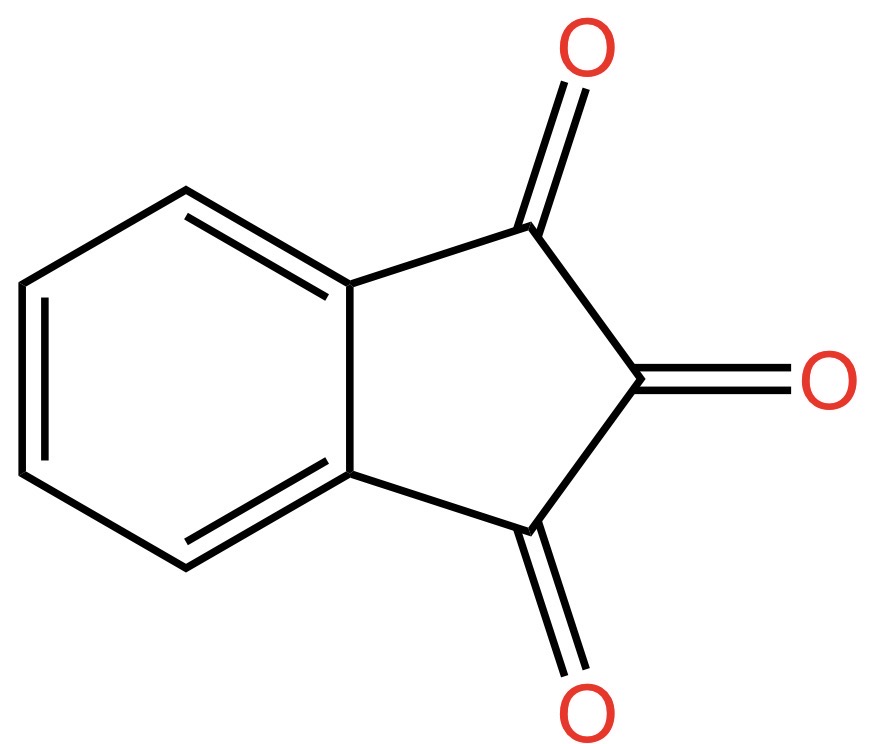
La reazione che produce questo oggetto chimico si chiama ossidazione, ma non importa il nome e proseguiamo. Andiamo avanti aggiungendo che i due gruppi ossigenati così ottenuti vengono indicati come “diosso”. Da qui il termine diossoidrindene. Ma dal momento che gli atomi di carbonio legati con doppio legame con l’ossigeno sono in posizione 1 e 3, il nome corretto è 1,3-diossoidrindene.
Continuiamo a mettere atomi di ossigeno e piazziamo due gruppi -OH, che si chiamano ossidrile oppure idrossile o, ancora, idrossi, sul carbonio in posizione 2. Otteniamo la struttura indicata nella seguente figura:
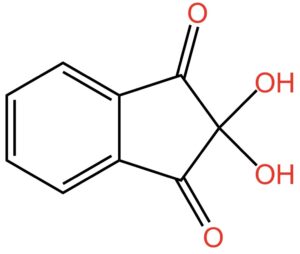
Poiché gli ossidrili in posizione 2 sono due, il nome dell’oggetto mostrato nella figura sopra diventa: 2,2-diidrossi-1,3-diossoidrindene… per gli amici intimi: ninidrina.
Quindi riassumendo
2,2-diidrossi è il suffisso che usiamo per indicare due gruppi -OH legati entrambi all’atomo di carbonio in posizione 2. Poiché gli ossidrili sono due, il numero civico del carbonio va ripetuto due volte mentre il suffisso “di” davanti a “idrossi” si riferisce proprio al numero di gruppi -OH presenti.
1,3-diosso è il suffisso che usiamo per indicare due atomi di ossigeno legati con doppio legame agli atomi di carbonio ai civici 1 e 3.
Il suffisso “idr” si riferisce a un sistema chimico che ha perso un doppio legame per effetto della reazione con idrogeno.
Infine, indene è la molecola da cui siamo partiti.
Ed ora?
Ed ora diamo un’occhiata al carbonio al civico 2 che è legato a due gruppi ossidrili.
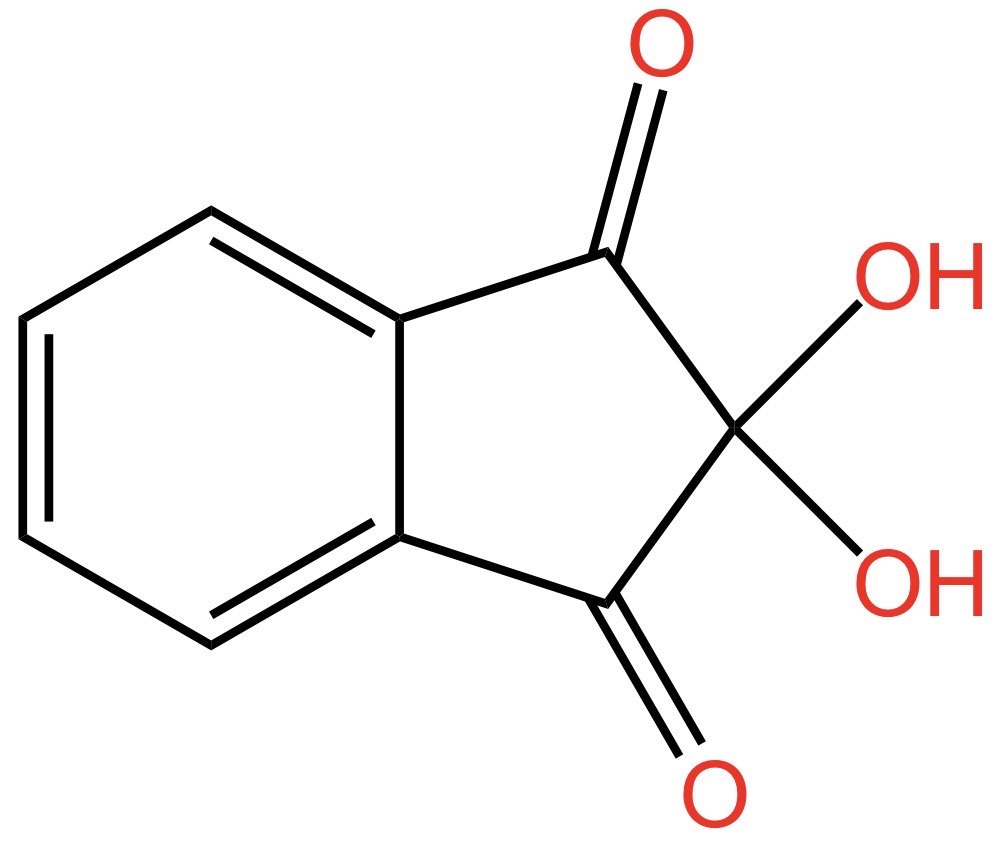
Dovete sapere che una molecola che contiene un gruppo ossidrile si chiama alcol. Sì… nel linguaggio comune il termine si riferisce all’ingrediente più importante di tante bevande: dal vino al bloody Mary, ovvero l’alcol etilico. Ma, in chimichese il termine alcol si riferisce a una classe di molecole che contengono il gruppo -OH. Quando nella stessa molecola sono presenti due gruppi ossidrili, questa viene indicata come diolo. Se i gruppi ossidrili sono sullo stesso atomo di carbonio, come nel caso della ninidrina, si parla di dioli geminali. Geminale deriva da gemello. I due -OH sullo stesso atomo di carbonio sono gemelli e come fratelli tendono a litigare. In effetti, i dioli geminali sono instabili perché i due gruppi -OH sullo stesso atomo di carbonio non stanno bene assieme e fanno a cazzotti. Cosa succede? Tendono a reagire per formare un doppio legame C=O in posizione 2, come nella figura qui sotto.
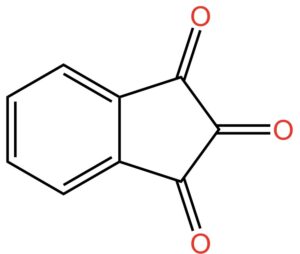
Adesso giochiamo ad aguzza la vista. Guardate cosa abbiamo combinato lì nella parte destra della molecola: ai civici da 1 a 3 ci sono tre gruppi C=O. Sembrano uguali, non è vero? Ma non è così.
Pur essendo uguali i tre gruppi hanno stabilità differente, o, potremmo anche dire, hanno reattività differente. In altre parole, tendono a reagire in modo diverso quando sono in presenza di particolari reagenti che andremo a vedere fra poco.
Il ruolo dell’elettronegatività
Il titolo di questo paragrafetto riporta una parolaccia: elettronegatività. Ma cos’è ‘sta roba?
Dobbiamo ricordarci che il chimichese è un linguaggio col proprio alfabeto e con le proprie regole di grammatica e sintassi. L’alfabeto è la tavola periodica. Essa contiene tutte le lettere, gli elementi chimici, che combinate formano le parole. Queste, a loro volta, messe assieme portano alle frasi chimiche che noi chimici ci ostiniamo a chiamare reazioni.
Sembra semplice, ma non lo è, purtroppo. E meno male… direi… altrimenti che ci sto a fare io qui?
Una delle regole grammaticali che a noi chimici piace utilizzare per spiegare cose che altrimenti sarebbero incomprensibili è l’elettronegatività. Il termine si riferisce alla tendenza di un elemento chimico a trattenere gli elettroni. Sì… proprio quegli affarini che si muovono intorno al nucleo secondo delle modalità che non ci è dato conoscere e che si presentano in delle regioni di spazio che amiamo definire col nome esotico di orbitali.
Più alta è la capacità di un elemento di trattenere gli elettroni, maggiore è il valore numerico dell’elettronegatività.
Sapete che il primo a quantificare l’elettronegatività è stato Linus Pauling? È uno dei miei idoli assieme a Richard Feynman. Li adoro entrambi.
Nella scala Pauling, l’elemento più elettronegativo è il fluoro (F), a seguire l’ossigeno. Solo molto dopo arriva il carbonio.
Quando scriviamo qualcosa come C-O, oppure C-H, stiamo riportando una interazione, che in chimichese si chiama legame, tra due elementi: carbonio e ossigeno, nel primo caso, carbonio e idrogeno, nel secondo.
Il trattino tra i due elementi è la rappresentazione grafica del legame chimico. Esso coinvolge due elettroni. In altre parole, il trattino sta a indicare due elettroni coinvolti nell’interazione tra gli elementi considerati.
Detto questo torniamo all’elettronegatività. Abbiamo detto che l’ossigeno è più elettronegativo del carbonio. Quindi, per effetto di questo, accade qualcosa di speciale nella distribuzione degli elettroni del legame C-O.
Siete curiosi di sapere cosa significa in un linguaggio meno chimichese?
Carbonio e ossigeno sono due persone. Il primo è un signore distinto col monocolo e il bastone; un signore d’altri tempi. Il secondo è un energumeno palestrato con la fissa per il wrestling. I due decidono di giocare al tiro alla fune. Chi vince secondo voi? Sì… l’energumeno palestrato.
Ecco… l’ossigeno, in virtù della sua maggiore elettronegatività, si comporta come il palestrato tirando verso di sé la corda con forza maggiore rispetto a quella usata dal signore di altri tempi. Ritornando al chimichese, gli elettroni del legame carbonio-ossigeno passano più tempo vicino a quest’ultimo che al carbonio. Nel linguaggio chimico più forbito, quello che userebbe il signore di altri tempi, la densità elettronica è spostata verso l’ossigeno. Questo vuol dire che, mediamente nel tempo, il carbonio è più elettropositivo dell’ossigeno. Scusate… traduco: per effetto della maggiore elettronegatività dell’ossigeno, il carbonio presenta una deficienza (nel senso di mancanza) elettronica ed ha una parziale carica positiva. Al contrario, l’ossigeno per gli stessi motivi ha una parziale carica negativa.
Adesso allarghiamo il campo. Se l’ossigeno richiama a sé gli elettroni del legame C-O, il carbonio, per effetto della sua deficienza elettronica, richiama a sé gli elettroni dei legami contigui. Quindi, se consideriamo qualcosa come C-C-O, anche il carbonio più lontano dall’ossigeno ha una parziale carica positiva indotta dal richiamo elettronico del lontano ossigeno.
Complichiamoci maggiormente l’esistenza e consideriamo un sistema come: O-C-C-C-O. Qui abbiamo tre atomi di carbonio e due di ossigeno. Gli atomi di carbonio alle estremità sono, ognuno, legati ad un atomo di ossigeno più elettronegativo. Questo vuol dire che il carbonio centrale risente del richiamo di due atomi di ossigeno. Come conseguenza, ha un potenziale elettropositivo (cioè una parziale carica positiva) maggiore rispetto al medesimo atomo di carbonio del sistema C-C-O.
Torniamo alla Ninidrina
Fatta la necessaria premessa sull’elettronegatività, riprendiamo la struttura della ninidrina in cui i due gruppi -OH hanno fatto a cazzotti. Per semplicità la riporto di nuovo qui di seguito:
Come vedete, abbiamo un C=O centrale, quello al civico 2, che risente dell’effetto dei due C=O al civico 1 e al civico 3. Pur avendo la stessa natura chimica, a causa degli effetti descritti prima, i C=O al civico 1 e 3 sono meno reattivi del C=O al civico 2. In pratica, la carica parziale netta positiva sul carbonio in 2 è maggiore rispetto a quelle degli atomi di carbonio in 1 e 3.
Cosa accade adesso?
Stiamo passeggiando nel parco. Stiamo per gli affari nostri quando incrociamo la nostra anima gemella: è amore a prima vista.
Ecco… il carbonio 2 della ninidrina povero di elettroni incrocia un sistema chimico che, invece, ne è ricco. Quale? Uno come R-NH2, per esempio. Questa è la formula generale dei composti che si chiamano ammine. L’azoto (N) è ricco di elettroni. Ne ha talmente tanti che quando si avvicina alla ninidrina viene attratto dalla sua anima gemella, ovvero il carbonio povero di elettroni in posizione 2. E cosa accade?
Guardate la figura qui sotto. In particolare la parte A. Qui le due anime gemelle si incontrano e si legano indissolubilmente. Le frecce curve indicano il movimento di elettroni dal sistema chimico che ne contiene di più a quello che ne contiene di meno. Si forma la molecola descritta nella parte B della medesima figura. In questa molecola intermedia, l’azoto è ancora ricco di elettroni. Di conseguenza li usa per rendere ancora più indissolubile il legame con la sua anima gemella portando alla formazione del composto riportato nella parte C della figura.
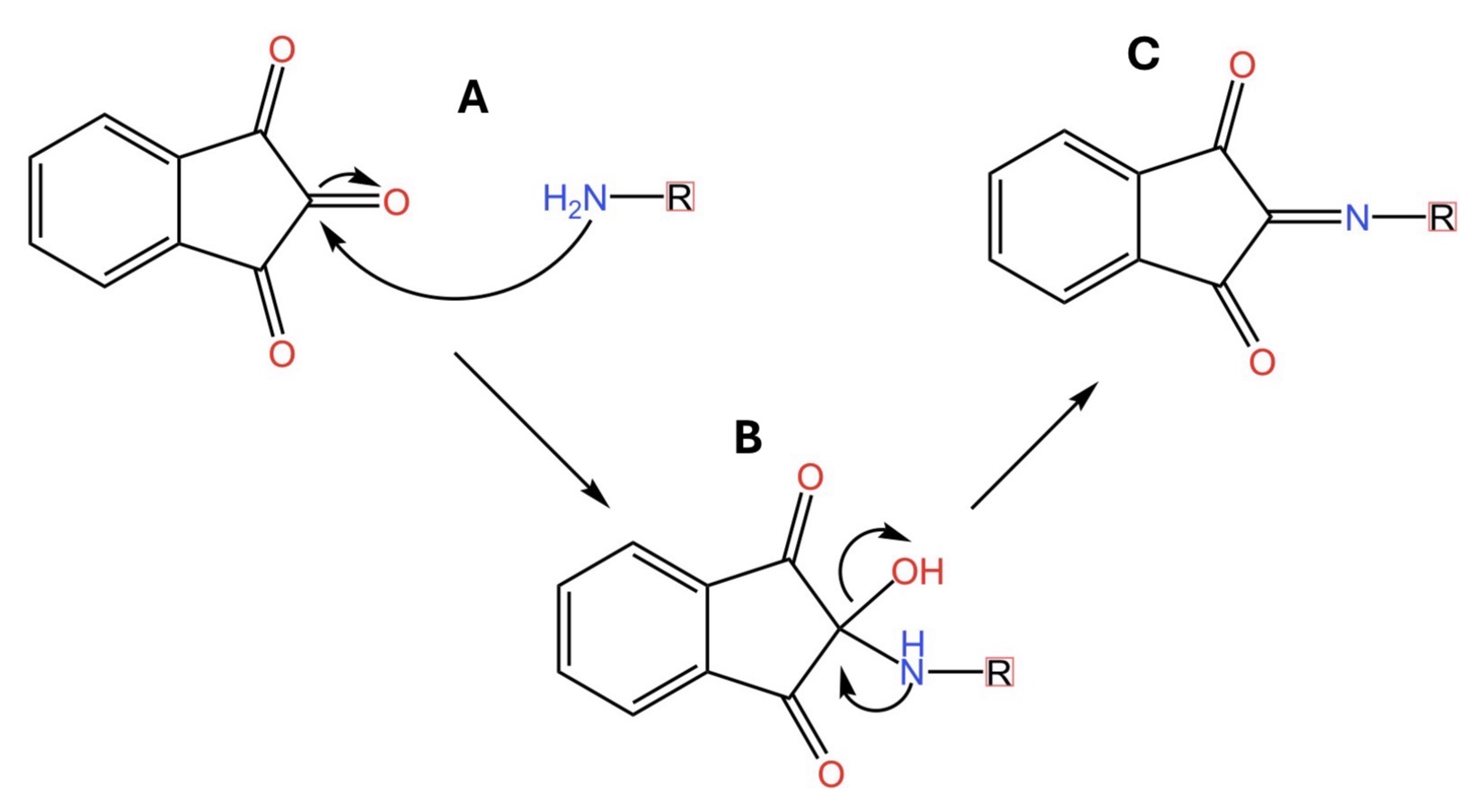
Sapete cosa è quella molecola? È un cromoforo, ovvero una molecola in grado di colorare di violetto una soluzione che la contiene. In altre parole, quando la ninidrina viene a contatto con una ammina o un altro sistema contenente un gruppo amminico, pensiamo, per esempio, agli amminoacidi, avviene una reazione che porta ad una soluzione colorata. L’intensità del colore è direttamente proporzionale alla quantità di sistema amminico che reagisce con la ninidrina. Nell’ambito della chimica biologica, la ninidrina è un utile strumento per quantificare gli amminoacidi con gruppi amminici primari (cioè del tipo RNH2) in sistemi proteici complessi.
La ninidrina nell’analisi forense
E veniamo al perché vi ho raccontato della ninidrina.
È da un po’ di tempo che si sente parlare del caso di Garlasco e dell’arresto quantomeno strano di Stasi. I diversi commentatori sui vari canali presenti in rete parlano ripetutamente di test della ninidrina per il rilievo delle impronte digitali. Ecco… ho l’impressione, ma è solo una mia opinione, che tutti parlino di qualcosa che non conoscono.
Cosa sono le impronte digitali? Si tratta di “immagini” dei nostri polpastrelli generate dal contatto con mezzi più o meno porosi. Da un punto di vista chimico, queste impronte contengono acqua, sali minerali, vitamine, amminoacidi e altro. Per il rilievo delle impronte si usa una soluzione di ninidrina che, reagendo con gli amminoacidi secondo lo schema appena descritto, produce un’immagine colorata che può essere studiata per consentire l’identificazione della persona che ha toccato quel dato oggetto.
Conclusioni
Perché la ninidrina mi divertiva quando studiavo la biochimica? Beh… da tutto quanto ho scritto è chiaro che conoscere la ninidrina significa avere in mente una quantità di conoscenze chimiche non indifferente. Non sto parlando solo della nomenclatura. Quella è una banalità. Bisogna avere conoscenze approfondite di chimica generale, chimica organica e quantomeccanica. Quello che vi ho raccontato non lo trovate scritto nei libri, ma lo ottenete dalla conoscenza comparata di diverse discipline chimiche. Ecco perché, quando sento qualcuno parlare della ninidrina come fosse un semplice reagente, sorrido. Per me è stata molto di più: è stata la prova che la chimica non è un insieme di nozioni, ma un modo di vedere il mondo. E quel viola, in provetta, era la conferma che avevo cominciato a vederlo davvero.
Grazie a tutti voi che avete avuto la pazienza di condividere con me un istante della mia vita da studente.












{kind=link}