
Cos’è la malattia di Huntington
La còrea o malattia di Huntington è una malattia genetica neurodegenerativa che colpisce la coordinazione muscolare e porta a un declino cognitivo e a problemi psichiatrici [1].
Non sono un neuroscienziato e non ho la pretesa di comprenderne fino in fondo le implicazioni biologiche e cliniche, che sono spiegate molto meglio altrove [2,3]. Ne parlo qui perché ho letto un interessantissimo articolo della senatrice Elena Cattaneo [4], che riesce a descrivere con grande chiarezza (anche per chi, come me, non è esperto di genetica) le basi molecolari della malattia.
La tripletta CAG e la proteina huntingtina
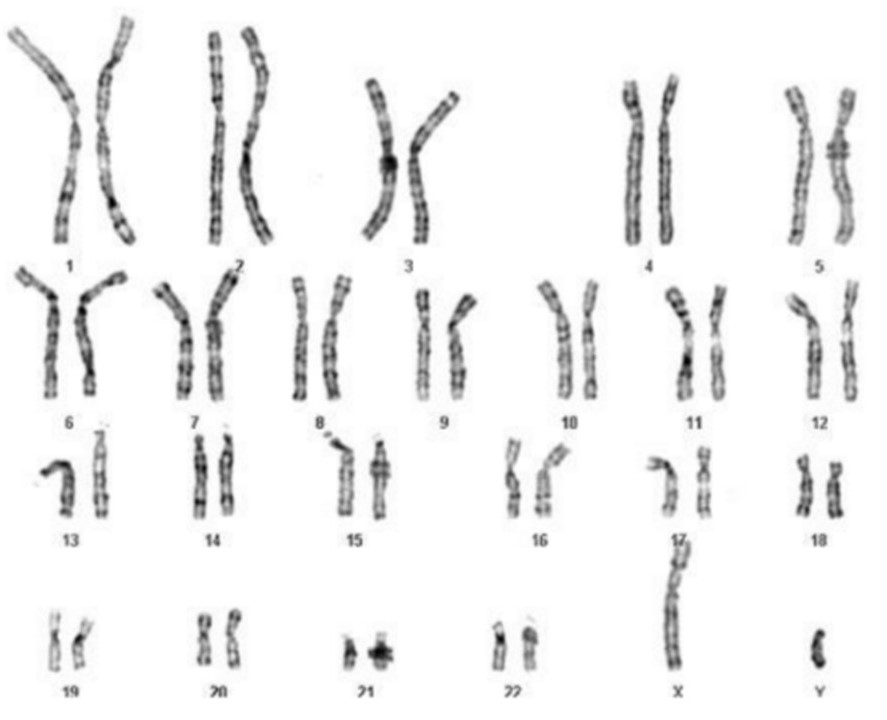
La malattia di Huntington è legata a un’anomalia di un gene localizzato sul cromosoma 4 (Figura 1), dove si trova una sequenza ripetuta della tripletta CAG.
Per i non addetti ai lavori: la tripletta CAG corrisponde a tre nucleotidi – citosina (C), adenina (A) e guanina (G) – e codifica per l’amminoacido glutammina (GLN) [5]. Sul cromosoma 4 questa tripletta si ripete molte volte (CAGCAGCAG…) e la sequenza codifica per una proteina che contiene un tratto con molte glutammine legate in fila.
La differenza sta proprio nel numero di ripetizioni:
- meno di 36 triplette → la proteina funziona normalmente e non si sviluppa la malattia;
- più di 36 triplette → la proteina subisce alterazioni strutturali che portano alla comparsa della malattia di Huntington.
Quando la sequenza è nella norma, la proteina stimola la produzione di una neurotrofina fondamentale per i neuroni striatali [4]. Se le ripetizioni superano la soglia critica, la produzione di questa neurotrofina può ridursi anche del 50% [4], causando gravi conseguenze per le cellule nervose.
Il dilemma evolutivo
A questo punto emerge la domanda cruciale: perché l’evoluzione ha conservato una sequenza così rischiosa, che nel tempo può diventare una condanna? [4]
Per rispondere, i ricercatori hanno dovuto fare un salto indietro nel tempo di circa 800 milioni di anni, fino ai primi organismi pluricellulari comparsi sulla Terra, come l’ameba sociale Dictyostelium [6]. Proprio lì è stato identificato il gene che, molto più tardi, nell’uomo, sarebbe stato associato all’Huntington.
Un vantaggio nascosto
La spiegazione suggerita è che questa sequenza ripetuta abbia avuto – e abbia ancora – un vantaggio evolutivo: nelle giuste quantità, la proteina che ne deriva sostiene lo sviluppo e la sopravvivenza dei neuroni, offrendo un beneficio che ha superato, almeno in termini evolutivi, il costo del rischio che le ripetizioni diventino eccessive.
Negli ultimi anni si è rafforzata l’idea che le ripetizioni CAG in numero non patologico non siano affatto neutre: studi su popolazioni sane hanno mostrato che variazioni nella lunghezza della sequenza, pur restando sotto la soglia critica, sono associate a differenze nelle strutture cerebrali e talvolta a prestazioni cognitive migliori [7,8]. Questo suggerisce che la sequenza poliglutaminica del gene HTT abbia un ruolo positivo nella plasticità e nello sviluppo del cervello.
Le espansioni somatiche
Un altro aspetto emerso di recente è quello delle espansioni somatiche: nel corso della vita, alcune cellule accumulano lentamente ulteriori ripetizioni CAG, senza che questo causi danni immediati. Tuttavia, quando la soglia viene superata in specifiche regioni cerebrali vulnerabili, il processo degenerativo si accelera bruscamente [9,10]. Questo meccanismo spiega perché la malattia si manifesta spesso in età adulta, dopo decenni di apparente normalità.
La prospettiva evolutiva
Analisi comparative in diverse specie hanno mostrato che la lunghezza della sequenza poliglutaminica nel gene HTT è stata oggetto di selezione naturale, a indicare che versioni con una certa estensione sono state probabilmente favorevoli allo sviluppo di funzioni nervose più complesse [11].
In altre parole, ciò che oggi leggiamo come malattia – la Huntington – potrebbe essere il rovescio della medaglia di un meccanismo che, nel corso dell’evoluzione, ha contribuito a plasmare la complessità del cervello umano, offrendo benefici cognitivi e adattativi a fronte di un rischio che si manifesta solo in alcune circostanze.
Riferimenti
[1] https://it.wikipedia.org/wiki/Malattia_di_Huntington
[2] http://www.uniroma2.it/didattica/MBND/deposito/lezione3.pdf
[3] http://www.telethon.it/…/malattie-tr…/huntington-malattia-di
[4] E. Cattaneo, Alla ricerca del gene perduto, Wired (Estate 2016), 77, 60-65
[5] https://www.ncbi.nlm.nih.gov/books/NBK545250/
[6] https://it.wikipedia.org/wiki/Dictyostelium
[7] Schultz JL, et al. Association of CAG Repeat Length in the Huntington Gene With Cognitive Performance in Young Adults. JAMA Neurology (2021).
[8] Cattaneo E, et al. When repetita no-longer iuvant: somatic instability of the CAG triplet in Huntington’s disease. Nucleic Acids Research (2025).
[9] Handsaker RE, et al. Long somatic DNA-repeat expansion drives neurodegeneration in Huntington’s disease. Cell (2025).
[10] Scahill RI, et al. Somatic CAG repeat expansion in blood associates with biomarkers of neurodegeneration in Huntington’s disease decades before clinical motor diagnosis. Nature Medicine (2025).
[11] Iennaco R, et al. The evolutionary history of the polyQ tract in huntingtin sheds light on its functional pro-neural activities. Cell Death & Differentiation (2022).
